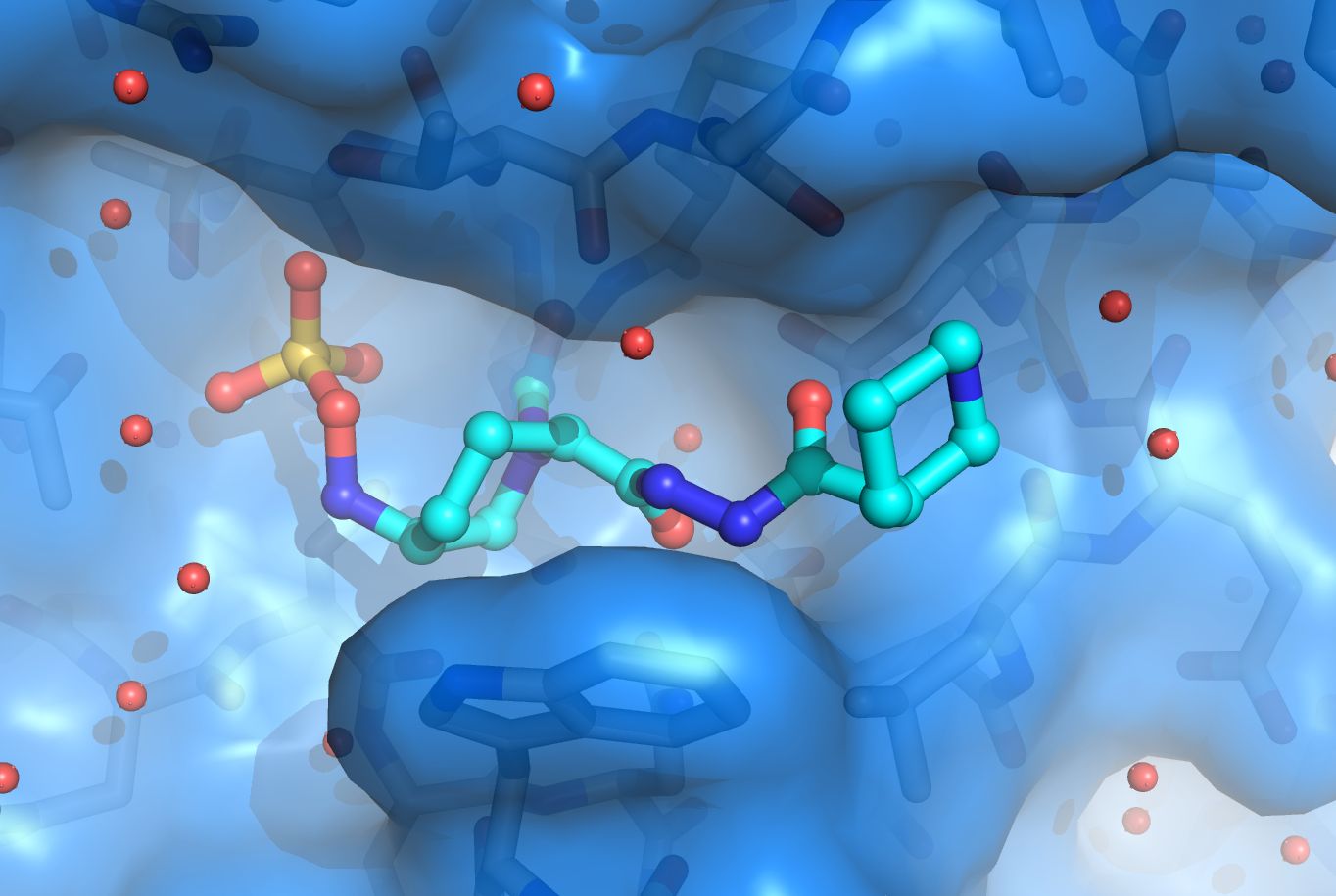
β-lactamases
Antibiotic resistance is a large clinical problem and in large part due to β-lactamases. The β-lactamases are present in the periplasm of Gram-negative bacteria and inactivate β-lactam antibiotics such as penicillin before they can reach their intended target to kill bacteria, the penicillin-binding proteins (PBPs). β-lactamases provide β-lactam antibiotic resistance to major pathogens such as Pseudomonas aeruginosa , Escherichia coli , Klebsiella pneumoniae , Acinetobacter baumannii , etc. The inhibition of β-lactamases is therefore desired which has led to the development of 6 clinically available β-lactamase inhibitors that are administered as a co-drug to potentiate β-lactam antibiotics. β-lactamases are, however, rapidly evolving and acquiring new clinically worrisome phenotypes ranging from being a carbapenemase (can inactivate the last-resort carbapenem β-lactams), or an extended-spectrum β-lactamase (ESBL; can inactivate cephalosporin β-lactams), or being inhibitor-resistant. The van den Akker lab is one of the leaders in the field studying β-lactamases structurally in collaboration with Dr. Robert Bonomo, M.D. and many others. Our lab has determined crystal structures of carbapenemases (such as the first published structure of KPC-2 β-lactamase, a clinically very important β-lactamase), ESBLs, inhibitor-resistant variants, studied the inhibition mechanism of many clinical and pre-clinical β-lactamase inhibitors, and carried out structure-based inhibitor design studies. β-lactamases currently under study include KPC-2, SHV-1, OXA-24/40, OXA-48, and PDC-3.
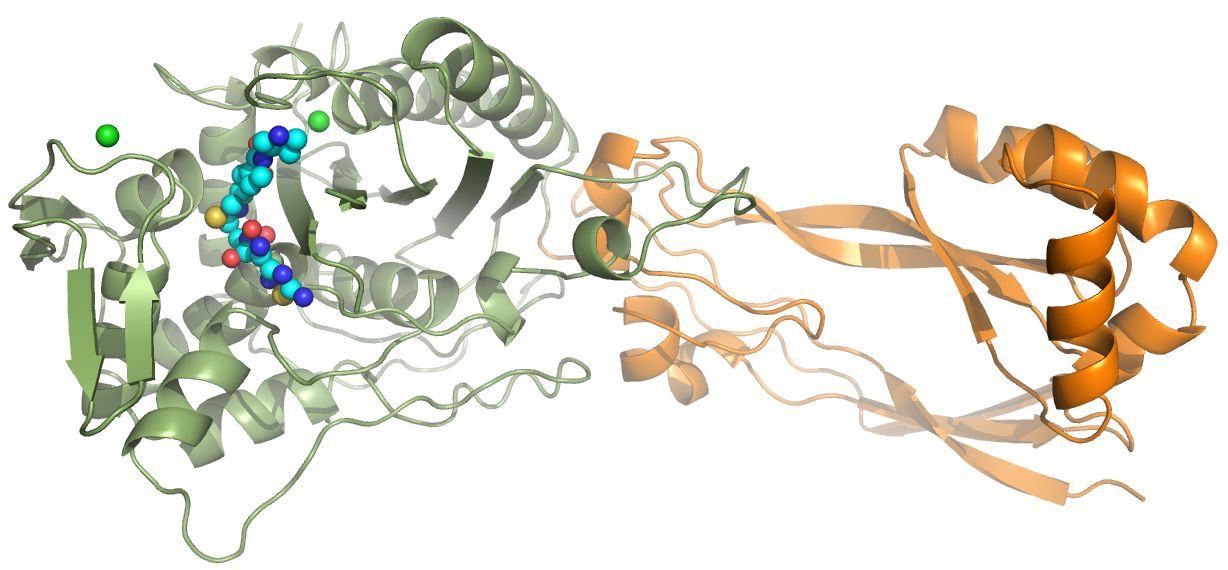
Penicillin-binding proteins (PBPs)
PBPs are responsible for the cross-linking of peptidoglycan strands to strengthen the cell wall of bacteria. PBP inhibition by for example the β-lactam penicillin can lead to bacterial cell death; β-lactams are, therefore, a very potent class of antibiotics. My lab studies the PBP3 of P. aeruginosa and we recently determined its structure in complex with ceftobiprole, a β-lactam marketed in several countries in and outside of Europe. Ceftobiprole is very active against certain key Gram-positive and Gram-negative bacteria; this work was done in collaboration with Basilea and Dr. Robert Bonomo (Kumar et al. 2020). We are also studying γ-lactam antibiotics which are resistant to degradation to β-lactamases (Goldberg et al, 2020). Finally, we also determined the crystal structure of a new PBP, that of P. aeruginosa PBP2 bound to WCK 5153. This inhibitor is a diazabicyclooctane inhibitor that also inhibits β-lactamases and is therefore a dual-target inhibitor or a β-lactam "enhancer" (Rajavel et al 2020).
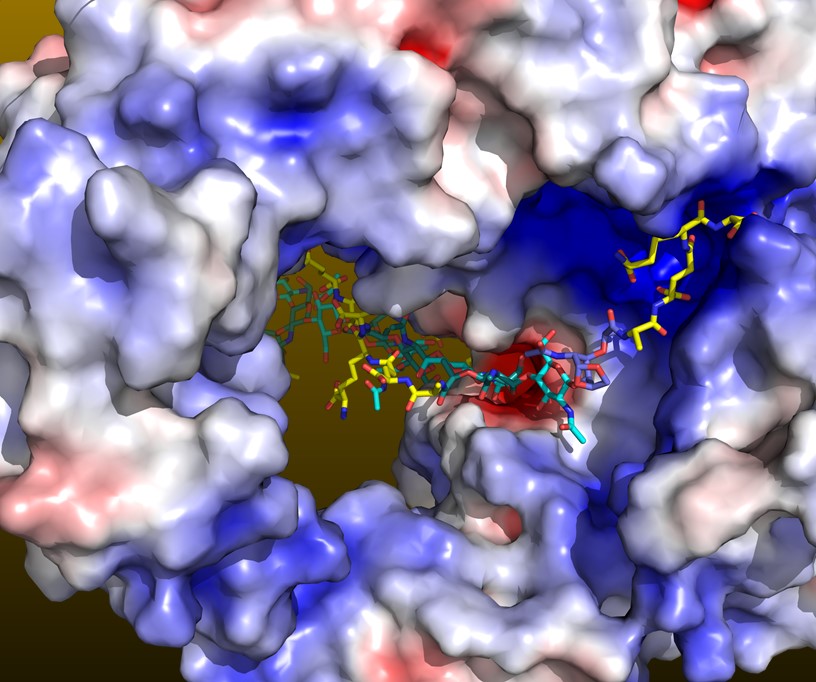
Lytic transglycosylases
Lytic transglycosylases (LTs) are bacterial enzymes that can hydrolyze peptidoglycan strands. These space making enzymes are needed to allow for example secretion systems or flagella to traverse through the peptidoglycan layer. In addition, LT function is needed during bacterial growth and cell division. Degradation products of LT can be recycled by bacteria and can lead to increases in β-lactamase expression, which is not desired when using β-lactam antibiotics to treat patients with infections. The inhibition of LTs is, therefore, a possible new approach to potentiate β-lactam antibiotic, furthermore aided by that the inhibition of both LTs and PBPs has been shown to lead to long uncross-linked peptidoglycan strands that weaken the cell wall. Our lab determined the structure of the Campylobacter jejuni LT with and without the natural product bulgecin A inhibitor bound (Vijayaraghavan et al. 2018). C. jejuni is a major foodborne pathogen. Amongst older adults >65 years, Campylobacter is the 2nd highest cause of hospitalization by bacterial enteric pathogens. We have an active fragment-based ligand discovery effort to develop novel LT inhibitors that could lead to novel treatments against C. jejuni and other pathogens.